
基因集富集分析(GeneSet Enrichment Analysis, GSEA),一种对基因进行富集分析的方法,即检验已知功能的基因集(即gene set,可以是从GO/KEGG/hallmark/MsigDB中拎出的某一特定类别的基因集合,也可以是自定义的功能基因集合),在一个依据与表型的相关度进行排序的基因列表(即两组样品的表达谱数据,依据基因在两种表型中的表达量的高低进行排序,因未对基因进行显著差异的筛选,因而可以将全部基因与不同表型的相关性均考虑进去)中是随机排列还是主要集中在列表的顶部或底部。若研究的已知功能基因集是非随机分布的,则说明该已知功能基因集与表型相关,根据其基因的集中情况,则 可以推断出该已知的功能具体和哪种表型更为接近。
GSEA分析结果的解读。
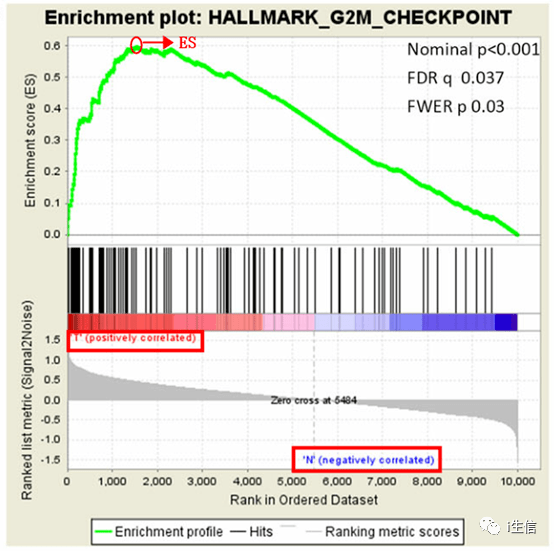 该图可分为3个部分:
(1) 上方绿色的EnrichmentScore折线图,横坐标为排序好的基因列表(即肿瘤组织和正常组织基因表达量矩阵表)。纵坐标是富集得分(Enrichment Score,ES).ES反映G2M功能相关基因集成员(HALLMARK_G2M_CHECKPOINT)在样品的基因表达排序列表的富集程度。折线图中的峰值就是这个基因集的ES值,ES值越高,说明样品在该通路中有富集。
(2) 中间部分每一条竖线代表功能基因集中的一个基因,及其在样本表型关联排序后的背景基因列表中的位置。存在显著富集的表型则与该已知的功能更为相关。
(3) 最下面为所有基因的rank值分布及信噪比,用以展示已知功能的基因在不同分组中的表达量情况,其中红色代表在“T”组(即肿瘤组)中高表达。从该图中可以看出,”HALLMARK_G2M_CHECKPOINT” 这个基因集是在“T”组(即肿瘤组)高表达的。
该图可分为3个部分:
(1) 上方绿色的EnrichmentScore折线图,横坐标为排序好的基因列表(即肿瘤组织和正常组织基因表达量矩阵表)。纵坐标是富集得分(Enrichment Score,ES).ES反映G2M功能相关基因集成员(HALLMARK_G2M_CHECKPOINT)在样品的基因表达排序列表的富集程度。折线图中的峰值就是这个基因集的ES值,ES值越高,说明样品在该通路中有富集。
(2) 中间部分每一条竖线代表功能基因集中的一个基因,及其在样本表型关联排序后的背景基因列表中的位置。存在显著富集的表型则与该已知的功能更为相关。
(3) 最下面为所有基因的rank值分布及信噪比,用以展示已知功能的基因在不同分组中的表达量情况,其中红色代表在“T”组(即肿瘤组)中高表达。从该图中可以看出,”HALLMARK_G2M_CHECKPOINT” 这个基因集是在“T”组(即肿瘤组)高表达的。
实战经验
- 注意目前GSEA broad官网上下载的gene set 都是human 的,所以如果对其他物种进行使用clusterprofiler进行分析,是会报错的,no mapping。 (Typically, GSEA uses gene sets from MSigDB. All gene sets in MSigDB consist of human gene symbols.)
- 如果需要做小鼠物种的GSEA 分析,则可以使用本网页中下载的 gene set数据集,这个网页中介绍了是采用何种方法,比如同源性匹配等,根据human 的gene set数据集,获得的小鼠的gene set数据集,可以用于做分析。这个网站下载的数据集是,rds格式的,并且读入之后,是list格式的,与clusterprofiler中GSEA函数需要的数据集格式不同,所以可以根据下面的代码进行对数据集整理。
c5 <- readRDS(file.path(pub_dir,
"Mm.c5.all.v7.1.entrez.rds"))
c5_gsea <- lapply(seq_along(c5), function(x)
c5[[x]] %>%
matrix(ncol = 1) %>%
as.data.frame() %>%
dplyr::rename(gene = V1) %>%
dplyr::mutate(term = names(c5)[x])) %>%
do.call(rbind,
args = .) %>%
dplyr::mutate(term = as.factor(term)) %>%
dplyr::select(term, gene)
致谢
| [看图说话 | GSEA分析–教你解锁高级的富集分析](https://www.sohu.com/a/439229098_652735) |